
ChIP-seq Analysis With R/Bioconductor
RとBioconductorでNGS解析: 3限 ChIP-seq データ解析
はじめに
この文章は統合データベース講習会:AJACSみちのく2「RとBioconductorを使ったNGS解析」3限目「ChIP-seq データ解析の基礎」の講義資料です。
この文章の著作権は二階堂愛にあります。ファイルのダウンロード、印刷、複製、大量の印刷は自由におこなってよいです。企業、アカデミアに関わらず講義や勉強会で配布してもよいです。ただし販売したり営利目的の集まりで使用してはいけません。ここで許可した行為について二階堂愛に連絡や報告する必要はありません。常に最新版を配布したいのでネット上での再配布や転載は禁止します。ネット上でのリンクはご自由にどうぞ。内容についての問い合わせはお気軽にメールしてください。
対象
ここでは、Rの基礎知識がある人を対象に、RとBioconductor を使った ChIP-seq データ解析について説明します。基本的な Unix コマンドが利用できることが前提です。
学習範囲
ChIP-seq を取り扱う。Peak解析については、Rではなく、ほかのオープンソースプログラムを利用して解析を行う。Rと Bioconductor ではその後の高次解析から論文の図の作成に至るまでを紹介します。
ChIP-seq
illumina HiSeq は、塩基とその精度を表す quality value のセットで出力します。これは fastq file と呼ばれており、すべての解析のスタートになります。
マッピング
まずこのファイルに含まれるシーケンスリードをリファレンスゲノムにマッピングすることで、どのゲノム領域由来のリードだったのかを調べます。ここでは、代表的なマッピングソフトである bowtie を用いて、マッピングを行います。
1 2 3 4 | |
各ディレクトリに *.sam というファイルができます。これはDNAショートリードとゲノム上の座標と保存するファイル形式です。これを bam file に変換します。
1 2 3 4 | |
変換された bam file を使って、binding site の場所を予測します。
1
| |
高次解析
準備
ここから R と Bioconductor を使っていきます。必要なパッケージをインストールします。まずRを起動します。
1
| |
Rに以下のコマンドを入力しパッケージをインストールします。
1 2 3 4 5 6 7 | |
以下のパッケージをダウンロードしインストールします。QuGAcomp
1
| |
最後にデータをダウンロードします。https://github.com/dritoshi/NGS_analysis_with_R_Bioconductor/tree/master/doc/results から *.bed ファイルを保存しておきます。
これで準備は終了です。
ピークデータの操作
ChIP-seq のデータは、区間データと呼ばれるデータ構造です。区間データとは、数直線上の2つの座標からなるデータです。ChIP-seq のピーク解析で得られたデータは、あるゲノムのある領域(区間)にタンパク質結合があるかどうかを示すデータ構造になっています。
MACS の場合は、bed file 形式の区間データが出力されます。このデータをRで読み込んでみます。
library("ChIPpeakAnno")
oct4.df <- read.table("results/Oct4_peaks.bed", header = FALSE)
sox2.df <- read.table("results/Sox2_peaks.bed", header = FALSE)
nanog.df <- read.table("results/Nanog_peaks.bed", header = FALSE)
is(oct4.df)## [1] "data.frame" "list" "oldClass"
## [4] "data.frameOrNULL" "vector"head(oct4.df)## V1 V2 V3 V4 V5
## 1 chr1 6448151 6448293 MACS_peak_1 11.91
## 2 chr1 7037487 7037628 MACS_peak_2 14.86
## 3 chr1 7303701 7303804 MACS_peak_3 14.42
## 4 chr1 7722943 7723046 MACS_peak_4 6.29
## 5 chr1 12734705 12734815 MACS_peak_5 8.33
## 6 chr1 12734855 12734958 MACS_peak_6 3.66oct4.gr <- BED2RangedData(oct4.df, header = FALSE)
sox2.gr <- BED2RangedData(sox2.df, header = FALSE)
nanog.gr <- BED2RangedData(nanog.df, header = FALSE)
is(oct4.gr)## [1] "RangedData" "DataTable" "List" "DataTableORNULL"
## [5] "Vector" "Annotated"head(oct4.gr)## RangedData with 6 rows and 2 value columns across 21 spaces
## space ranges | strand score
## <factor> <IRanges> | <numeric> <numeric>
## MACS_peak_1 1 [ 6448151, 6448293] | 1 11.91
## MACS_peak_2 1 [ 7037487, 7037628] | 1 14.86
## MACS_peak_3 1 [ 7303701, 7303804] | 1 14.42
## MACS_peak_4 1 [ 7722943, 7723046] | 1 6.29
## MACS_peak_5 1 [12734705, 12734815] | 1 8.33
## MACS_peak_6 1 [12734855, 12734958] | 1 3.66ファイルからいったん data frame として bed file を読み込みます。それを、BED2RangedData を用いて RangedData オブジェクトに変換します。このデータ構造にしておくことで、Bioconductor が提供する様々な区間計算やバイオインフォマティクスの機能が使えるようになります。
ピークデータのアノテーション
それぞれのピークがどの遺伝子の側にあるのかを計算します。TSS (transcript start site) からのピークまでの距離を計算します。
data(TSS.mouse.NCBIM37)
oct4.anno <- annotatePeakInBatch(RangedData(oct4.gr), AnnotationData = TSS.mouse.NCBIM37,
output = "both")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")## Warning: 'matchMatrix' is deprecated. Use 'as.matrix' instead. See
## help("Deprecated")is(oct4.anno)## [1] "RangedData" "DataTable" "List" "DataTableORNULL"
## [5] "Vector" "Annotated"head(oct4.anno)## RangedData with 6 rows and 9 value columns across 21 spaces
## space ranges |
## <factor> <IRanges> |
## MACS_peak_10 ENSMUSG00000025927 1 [ 19234791, 19234953] |
## MACS_peak_101 ENSMUSG00000026698 1 [163899326, 163899435] |
## MACS_peak_102 ENSMUSG00000026585 1 [165709520, 165709737] |
## MACS_peak_103 ENSMUSG00000041396 1 [165950038, 165950155] |
## MACS_peak_104 ENSMUSG00000026575 1 [166307302, 166307438] |
## MACS_peak_106 ENSMUSG00000000544 1 [168065279, 168065422] |
## peak strand
## <character> <character>
## MACS_peak_10 ENSMUSG00000025927 MACS_peak_10 +
## MACS_peak_101 ENSMUSG00000026698 MACS_peak_101 +
## MACS_peak_102 ENSMUSG00000026585 MACS_peak_102 +
## MACS_peak_103 ENSMUSG00000041396 MACS_peak_103 +
## MACS_peak_104 ENSMUSG00000026575 MACS_peak_104 +
## MACS_peak_106 ENSMUSG00000000544 MACS_peak_106 +
## feature start_position
## <character> <numeric>
## MACS_peak_10 ENSMUSG00000025927 ENSMUSG00000025927 19198995
## MACS_peak_101 ENSMUSG00000026698 ENSMUSG00000026698 163899319
## MACS_peak_102 ENSMUSG00000026585 ENSMUSG00000026585 165709810
## MACS_peak_103 ENSMUSG00000041396 ENSMUSG00000041396 165925018
## MACS_peak_104 ENSMUSG00000026575 ENSMUSG00000026575 166237803
## MACS_peak_106 ENSMUSG00000000544 ENSMUSG00000000544 168060591
## end_position insideFeature
## <numeric> <character>
## MACS_peak_10 ENSMUSG00000025927 19228815 downstream
## MACS_peak_101 ENSMUSG00000026698 163903566 inside
## MACS_peak_102 ENSMUSG00000026585 165847231 upstream
## MACS_peak_103 ENSMUSG00000041396 165927374 downstream
## MACS_peak_104 ENSMUSG00000026575 166334805 inside
## MACS_peak_106 ENSMUSG00000000544 168096640 inside
## distancetoFeature shortestDistance
## <numeric> <numeric>
## MACS_peak_10 ENSMUSG00000025927 35796 5976
## MACS_peak_101 ENSMUSG00000026698 7 7
## MACS_peak_102 ENSMUSG00000026585 -290 73
## MACS_peak_103 ENSMUSG00000041396 25020 22664
## MACS_peak_104 ENSMUSG00000026575 69499 27367
## MACS_peak_106 ENSMUSG00000000544 4688 4688
## fromOverlappingOrNearest
## <character>
## MACS_peak_10 ENSMUSG00000025927 NearestStart
## MACS_peak_101 ENSMUSG00000026698 NearestStart
## MACS_peak_102 ENSMUSG00000026585 NearestStart
## MACS_peak_103 ENSMUSG00000041396 NearestStart
## MACS_peak_104 ENSMUSG00000026575 NearestStart
## MACS_peak_106 ENSMUSG00000000544 NearestStart遺伝子とピークの位置の関係をサマリーします。
oct4.anno.table <- table(as.data.frame(oct4.anno)$insideFeature)
is(oct4.anno.table)## [1] "table" "oldClass"head(oct4.anno.table)##
## downstream includeFeature inside overlapEnd overlapStart
## 406 1 702 7 74
## upstream
## 768可視化します。
barplot(oct4.anno.table)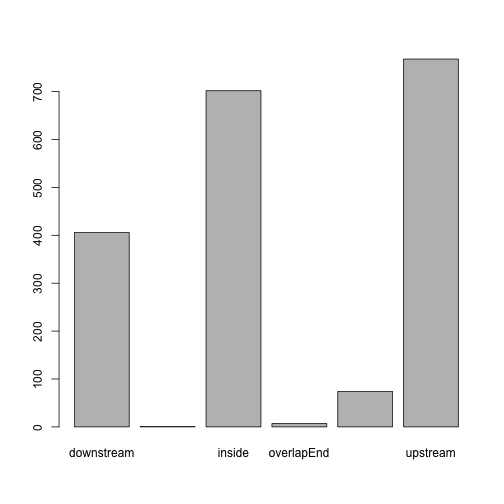
これをそれぞれの転写因子の ChIP-seq データで描画することで、結合領域の好みを知ることができます。pie を使えばパイチャートを描くこともできますが、今回のように、別々の転写因子のデータを比較する場合には、pie chart は使うべきではありません。人間は長さの比較に対して、面積の比較をするのが難しいためです。
次にピークがアサインされた遺伝子になんらかの特徴があるか見てみましょう。これは ChIP-seq の実験がうまくいっているかざっくりと評価するためにも重要です。ここでは頻出する Gene ontology を挙げます。
library(org.Mm.eg.db)
oct4.go <- getEnrichedGO(oct4.anno, orgAnn = "org.Mm.eg.db", maxP = 0.01, multiAdj = TRUE,
minGOterm = 10, multiAdjMethod = "BH")
oct4.bp.goterm <- unique(oct4.go$bp[order(oct4.go$bp[, 10]), c(2, 10)])
oct4.cc.goterm <- unique(oct4.go$cc[order(oct4.go$cc[, 10]), c(2, 10)])
oct4.mf.goterm <- unique(oct4.go$mf[order(oct4.go$mf[, 10]), c(2, 10)])
is(oct4.mf.goterm)## [1] "data.frame" "list" "oldClass"
## [4] "data.frameOrNULL" "vector"head(oct4.mf.goterm)## go.term
## 134 nucleic acid binding transcription factor activity
## 710 sequence-specific DNA binding transcription factor activity
## 477 DNA binding
## 1324 sequence-specific DNA binding
## 48 sequence-specific DNA binding RNA polymerase II transcription factor activity
## 668 chromatin binding
## BH.adjusted.p.value
## 134 2.257e-07
## 710 2.257e-07
## 477 4.183e-06
## 1324 1.852e-05
## 48 3.147e-05
## 668 9.265e-05ピーク領域の配列を得る
ピーク領域のDNA配列を得ることは2つの意味で重要になります。ひとつは、その配列を用いて、PCRプライマーを設計し、ChIP産物に対して、ChIP-qPCR を行うことで、解析結果の評価が実験によって行うことができます。これはピーク解析のスコアの閾値を決定するうえでも重要です。
もうひとつはピークの配列のなかに頻出するDNA配列パターン、すなわち、DNAモチーフを発見するために必要となるからです。DNAモチーフを発見することで、タンパク質がどのようなDNA配列を認識して結合するのかを明らかにすることができます。またすでにモチーフ配列が既知の場合は、その含有率を知ることで、ChIP-seq の実験がうまくいっているかを評価することができます。
では配列を得てみます。
library("BSgenome.Mmusculus.UCSC.mm9")
oct4.peaksWithSeqs <- getAllPeakSequence(oct4.gr, upstream = 0, downstream = 0,
genome = Mmusculus)
is(oct4.peaksWithSeqs)## [1] "RangedData" "DataTable" "List" "DataTableORNULL"
## [5] "Vector" "Annotated"head(oct4.peaksWithSeqs)## RangedData with 6 rows and 4 value columns across 21 spaces
## space ranges | upstream downstream
## <factor> <IRanges> | <numeric> <numeric>
## MACS_peak_1 chr1 [ 6448151, 6448293] | 0 0
## MACS_peak_2 chr1 [ 7037487, 7037628] | 0 0
## MACS_peak_3 chr1 [ 7303701, 7303804] | 0 0
## MACS_peak_4 chr1 [ 7722943, 7723046] | 0 0
## MACS_peak_5 chr1 [12734705, 12734815] | 0 0
## MACS_peak_6 chr1 [12734855, 12734958] | 0 0
## sequence
## <character>
## MACS_peak_1 TTCTTTCCTCCTTTGTACCCTGGGCGCTATAGGAATTCAACTTTACAAGCTGTGAGGAAATGGTGATTCTTGTGCAAAGTGAACAGCTGGGTCTGTCAACAGAAGGTAGCATTCCTTTGATACTGAGCCTTCCTGGTGTGGCA
## MACS_peak_2 CTGCTAAGACCATTTGTGGCTTTAAGAGCCTCCTCTGCCTCCATTTACATATCTACTGTCTGCCTTCTGCCAAGCCACAGAGCAGTTTGAAGGACAGCTTCAGCATTCCTACAATACAGGGCTGTGATGTTGCATTGGTTCA
## MACS_peak_3 CAACTTTTGTTATTCATAGCAGAAGTCACACCAGTCCACAGCTGAATAGCCACAGTGTTAAGAACAGCTGTCTTACAGCACTGTATGTGGAGAACAGAAAGAGC
## MACS_peak_4 TTTAGGTGTAAAGGTACAGGAAGTCCTGTGGTGTGGTGATGAAGGGCACCATCTTTGAATATGAAAAGAGCTTTTGTAAAAGTCTGCAGGAGCTGATTTAAGGA
## MACS_peak_5 ACCTGGTTTGGGGGTGAAAGCACAGGAAGCCACTGCATTTTGCATAGTTTCCATTTGAAAAGGTCTACCAGCCCGGGTTCTCTTTCAGGGTGGGCCAGGCTTCCGGGAACC
## MACS_peak_6 TTTGTATCAGATCCTCAAATGCCACTCAGCTAGTTCCACCTTTGAAAAGCTGCCATTGTAATGTCCCCAGAGTGTGCATAGCCATTCCTAAAGGGTGAGGTGGA
## strand
## <numeric>
## MACS_peak_1 1
## MACS_peak_2 1
## MACS_peak_3 1
## MACS_peak_4 1
## MACS_peak_5 1
## MACS_peak_6 1write2FASTA(oct4.peaksWithSeqs, file = "results/oct4.peaksWithSeqs.fa")## Warning: 'writeFASTA' is deprecated. Use 'write.XStringSet' instead. See
## help("Deprecated")最終的には、FASTA 形式でファイルに保存しています。
ピーク領域に頻出するDNAモチーフを得る
では、前項で作成した配列に含まれるDNAモチーフを発見してみます。
library("rGADEM")
oct4.seqs <- read.DNAStringSet("results/oct4.peaksWithSeqs.fa", "fasta")
oct4.motif <- GADEM(oct4.seqs, verbose = 1, genome = Mmusculus)## *** Start C Programm ***is(oct4.seqs)## [1] "DNAStringSet" "XStringSet" "XRawList" "XVectorList"
## [5] "List" "Vector" "Annotated"head(oct4.seqs)## A DNAStringSet instance of length 6
## width seq names
## [1] 143 TTCTTTCCTCCTTTGTACCCT...TGAGCCTTCCTGGTGTGGCA MACS_peak_1
## [2] 142 CTGCTAAGACCATTTGTGGCT...TGTGATGTTGCATTGGTTCA MACS_peak_2
## [3] 104 CAACTTTTGTTATTCATAGCA...ATGTGGAGAACAGAAAGAGC MACS_peak_3
## [4] 104 TTTAGGTGTAAAGGTACAGGA...TGCAGGAGCTGATTTAAGGA MACS_peak_4
## [5] 111 ACCTGGTTTGGGGGTGAAAGC...GGGCCAGGCTTCCGGGAACC MACS_peak_5
## [6] 104 TTTGTATCAGATCCTCAAATG...TTCCTAAAGGGTGAGGTGGA MACS_peak_6is(oct4.motif)## [1] "gadem"head(oct4.motif)## Object of class 'gadem'
## This object has the following slots:
## motifs,pwm,consensus,align,name,seq,chr,start,end,strand,seqID,pos,pval,fastaHeader次は発見したモチーフが既知のモチーフに似ているかどうかを調べます。ここでは、JASPAR という転写因子結合モチーフのデータベースに対し、発見したモチーフに似たものを検索します。実際には、PWM の相関係数を計算しています。
library("MotIV")
## prep. database
path <- system.file(package = "MotIV")
jaspar <- readPWMfile(file.path(path, "extdata/jaspar2010.txt"))
jaspar.scores <- readDBScores(file.path(path, "extdata/jaspar2010_PCC_SWU.scores"))
## search motifs
oct4.motif.pwms <- getPWM(oct4.motif)
oct4.jaspar <- motifMatch(inputPWM = oct4.motif.pwms, align = "SWU", cc = "PCC",
database = jaspar, DBscores = jaspar.scores, top = 5)##
## Ungapped Alignment
## Scores read
## Database read
## Motif matches : 5## output results with sequence logos
plot(oct4.jaspar, ncol = 2, top = 5, rev = FALSE, main = "Oct4 ChIP-seq", bysim = TRUE)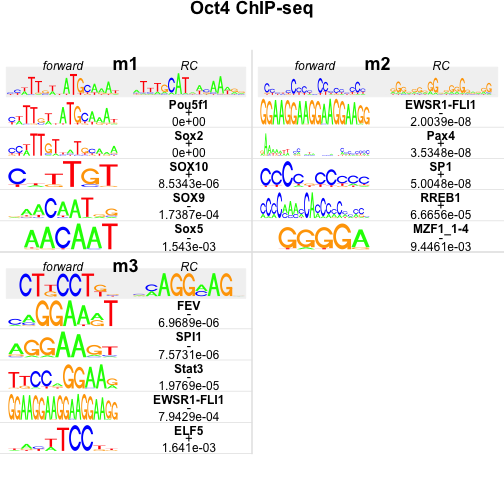
異なるChIP-seqデータの比較
異なる転写因子やエピジェネティクス因子の ChIP-seq のデータを比較したり、peak 解析ソフトによる結果の比較をしたい場合がある。このときは、それぞれの peak データを取り込んで、その重なりの数を数えるのが一般的である。
より定量的に比較するには、それぞれのピークの位置にどのぐらい相関があるかを計算する。まずピークの区間データを、ある座標に peka が存在するかどうかを 0, 1 で示した localization vector と呼ばれるデータに変換する。次に、適切な大きさに、区間ごとに binning (window analysis, smoothing) した後、そのベクター間の相関を計算する。
以下では3つの転写因子の peak データをピアソンの積率相関係数によって比較する方法である。これは 2 x 2 contingency table (cross table) の phi coefficient と等価である。一般的に、localization vector の相関は高くならないので注意が必要である。
library("QuGAcomp")## Loading required package: IRanges## Loading required package: BiocGenerics## Attaching package: 'BiocGenerics'## The following object(s) are masked from 'package:stats':
##
## xtabs## The following object(s) are masked from 'package:base':
##
## anyDuplicated, cbind, colnames, duplicated, eval, Filter, Find, get,
## intersect, lapply, Map, mapply, mget, order, paste, pmax, pmax.int, pmin,
## pmin.int, Position, rbind, Reduce, rep.int, rownames, sapply, setdiff,
## table, tapply, union, unique## Attaching package: 'IRanges'## The following object(s) are masked from 'package:plyr':
##
## compact, desc, rename## Loading required package: GenomicRangeslibrary("corrplot")
genome.length.file <- file.path(system.file(package = "QuGAcomp"), "data", "mm9.info")
oct4.bed.file <- file.path(system.file(package = "QuGAcomp"), "data", "GSM288346_ES_Oct4.mm9.header.bed")
sox2.bed.file <- file.path(system.file(package = "QuGAcomp"), "data", "GSM288347_ES_Sox2.mm9.header.bed")
nanog.bed.file <- file.path(system.file(package = "QuGAcomp"), "data", "GSM288345_ES_Nanog.mm9.header.bed")
oct4.gr <- loadBedFile(oct4.bed.file, genome.length.file)## [1] 3760 6sox2.gr <- loadBedFile(sox2.bed.file, genome.length.file)## [1] 4526 6nanog.gr <- loadBedFile(nanog.bed.file, genome.length.file)## [1] 10343 6oct4.fat <- fat(oct4.gr, 200)## Warning: trimmed end values to be <= seqlengthssox2.fat <- fat(sox2.gr, 200)
nanog.fat <- fat(nanog.gr, 200)
oct4.unistd <- unifyStrand(oct4.fat)
sox2.unistd <- unifyStrand(sox2.fat)
nanog.unistd <- unifyStrand(nanog.fat)
oct4.cov <- coverage(oct4.unistd)
sox2.cov <- coverage(sox2.unistd)
nanog.cov <- coverage(nanog.unistd)
oct4.bin500 <- lapply(oct4.cov, function(x) rleBinning(x, 500))
sox2.bin500 <- lapply(sox2.cov, function(x) rleBinning(x, 500))
nanog.bin500 <- lapply(nanog.cov, function(x) rleBinning(x, 500))
oct4.bin500 <- flatRleList(oct4.bin500)
sox2.bin500 <- flatRleList(sox2.bin500)
nanog.bin500 <- flatRleList(nanog.bin500)
quga.oct4.sox2 <- qugacomp(oct4.bin500, sox2.bin500)
quga.oct4.nanog <- qugacomp(oct4.bin500, nanog.bin500)
quga.sox2.nanog <- qugacomp(sox2.bin500, nanog.bin500)
num <- 3
mat.cor <- matrix(0, nrow = num, ncol = num)
rownames(mat.cor) <- c("Oct4", "Sox2", "Nanog")
colnames(mat.cor) <- c("Oct4", "Sox2", "Nanog")
diag(mat.cor) <- rep(1, num)
mat.cor[1, 2] <- mat.cor[2, 1] <- pearsonCoef(quga.oct4.sox2)
mat.cor[1, 3] <- mat.cor[3, 1] <- pearsonCoef(quga.oct4.nanog)
mat.cor[2, 3] <- mat.cor[3, 2] <- pearsonCoef(quga.sox2.nanog)
mat.cor.max <- max(mat.cor[upper.tri(mat.cor, diag = F)])
mat.cor.min <- min(mat.cor[upper.tri(mat.cor, diag = F)])
corrplot(mat.cor, method = "circle", type = "full", diag = FALSE, outline = FALSE,
addcolorlabel = "bottom", cl.lim = c(mat.cor.min, mat.cor.max), cl.ratio = 0.2)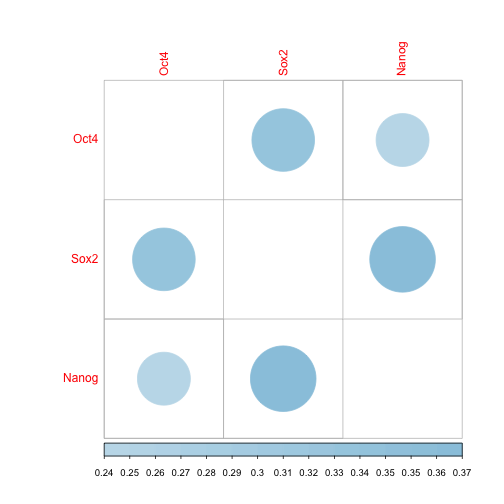
次に学ぶこと
ここでは、シーケンス実験のQCについては扱っていませんでした。ChIP-seq の場合はシーケンスライブラリを作成するときに、PCR 増幅をしますが、元々サンプルの準備が難しい実験ですので、PCRサイクルが多くなってしまう場合があります。また、ChIP assay 自体が抗体やサンプルによって、十分にタンパク質結合領域を enrich できない場合もあります。そのため、QCは非常に重要なステップになります。
特に、ChIP-qPCR によって enrichnment が確認された領域、DNA断片が落ちてこない領域などの情報を事前に入手し、その領域にリードがどの程度含まれるかを確認する必要があります。また、まったく同一のリードというのは確率的にあまり出てこないはずですが、これがどのぐらい含まれるかを定量することも大切です。もし PCR のサイクル数が過剰な場合は、この重複リード率が増える傾向にあります。
また、ChIP-seq は、RNA-seq よりもゲノムのリピート領域にマップされる遺伝子が多い傾向があります。このとき、リピート領域にマップされたリードが、真のタンパク質結合サイトに含まれるリードに比べて、かなり多くなります。そのため、peak 解析のスコアの計算に影響を与える場合があるので、マッピングが終了後に、これらを除くことが重要です。
このような問題点の指標になる数値やデータ操作、解析についても R/Bioconductor で計算することが可能です。このようなステップを入れることで、より正確な解析と実験へのフィードバックができるデータ解析パイプラインが作成することができるでしょう。
実行環境
sessionInfo()## R version 2.15.1 (2012-06-22)
## Platform: x86_64-apple-darwin9.8.0/x86_64 (64-bit)
##
## locale:
## [1] ja_JP.UTF-8/ja_JP.UTF-8/ja_JP.UTF-8/C/ja_JP.UTF-8/ja_JP.UTF-8
##
## attached base packages:
## [1] datasets utils stats graphics grDevices methods base
##
## other attached packages:
## [1] corrplot_0.60 QuGAcomp_0.99.2 GenomicRanges_1.8.12
## [4] IRanges_1.14.4 BiocGenerics_0.2.0 knitr_0.7.6
## [7] stringr_0.6.1 RColorBrewer_1.0-5 MASS_7.3-20
## [10] plyr_1.7.1 BiocInstaller_1.4.7
##
## loaded via a namespace (and not attached):
## [1] colorspace_1.1-1 dichromat_1.2-4 digest_0.5.2 evaluate_0.4.2
## [5] formatR_0.6 ggplot2_0.9.1 grid_2.15.1 labeling_0.1
## [9] memoise_0.1 munsell_0.3 proto_0.3-9.2 reshape2_1.2.1
## [13] scales_0.2.1 stats4_2.15.1 tools_2.15.1