
RNA-seq Analysis With R/Bioconductor
RとBioconductorでNGS解析: 2限 RNA-seq データ解析
はじめに
この文章は統合データベース講習会:AJACSみちのく2「RとBioconductorを使ったNGS解析」2限目「RNA-seq データ解析」の講義資料です。
この文章の著作権は二階堂愛にあります。ファイルのダウンロード、印刷、複製、大量の印刷は自由におこなってよいです。企業、アカデミアに関わらず講義や勉強会で配布してもよいです。ただし販売したり営利目的の集まりで使用してはいけません。ここで許可した行為について二階堂愛に連絡や報告する必要はありません。常に最新版を配布したいのでネット上での再配布や転載は禁止します。ネット上でのリンクはご自由にどうぞ。内容についての問い合わせはお気軽にメールしてください。
対象
ここでは、Rの基礎知識がある人を対象に、RとBioconductor を使った RNA-seq データ解析について説明します。基本的な Unix コマンドが利用できることが望ましいです。
学習範囲
RNA-seq を取り扱います。データのQCやマッピング、発現量の定量については、Rではなく、ほかのオープンソースプログラムを利用して解析を行います。Rと Bioconductor ではその後の高次解析から論文の図の作成に至るまでを紹介します。データについては Bioconductor のパッケージに添付されている RNA-seq データを使います。
RNA-seq
illumina HiSeq は、塩基とその精度を表す quality value のセットで出力します。これは fastq file と呼ばれており、すべての解析のスタートになります。
マッピング
まずこのファイルに含まれるシーケンスリードをリファレンスゲノムにマッピングすることで、どのゲノム領域由来のリードだったのかを調べます。RNA-seq の場合は、mRNA が splicing された後のシーケンスも含まれるので、それを考慮したマッピングアルゴリズムが必要になります。ここでは、spliced alignment に対応した代表的なマッピングソフトである tophat2 を用いて、マッピングを行います。
ここでは、paired-end で、paired-read 同士の距離が 100 bp であることを想定します。bowtie2_indexes/mm9 には、検索しやすいようファイル変換(インデックス化)されたゲノム配列を指定しています。ここではマウスゲノム mm9 を指定しています。ftp://ftp.cbcb.umd.edu/pub/data/bowtie_indexes/ からダウンロードできます。
その後、ペアの fastq を forward, reverse の順で指定しています。ファイル名は、それぞれ実験名1.fastq, 実験名2.fastq とします。-o では結果を出力するディレクトリを指定します。single-end の場合は、ひとつファイルを指定し、-r を除きます。
-p は計算に利用する CPU cores の数を整数でします。以下の例では、8 CPU cores あるコンピュータを使うので、8 を指定していますが、お使いのコンピュータに合せて変えてください。
1 2 3 4 5 6 7 8 | |
それぞれのディレクトリに accepted_hits.bam というファイルができます。これはゲノムの座標とショートリードを格納するバイナリファイルの形式で、非常によく使われる形式です。ちなみに、この bam file を操作するツールとして有名なのが samtools です。
発現量の定量
次にマッピングデータから発現量を定量します。出力された bam file を利用します。まず bam file を適切な名前に rename します。
1 2 3 4 5 6 7 8 9 | |
次に、cufflinks の cuffdiff コマンド使って、発現量の定量と発現差解析を行います。-p は CPU cores の数です。-L はサンプルのラベルで好きな文字列を指定できますが、今後の解析にそのまま利用し、作成される図にもそのまま表示されるものなので、慎重に選んでください。-o は結果を出力するディレクトリ名です。そのあとに、sample, control の順番にファイルを指定します。replication しているサンプルは、コンマで区切ります。sample と control の区切りはスペース文字であることに注意してください。ensembl_gene.gtf はリファレンスとなるトランスクリプトームの情報です。この GTF file はイルミナ社のウェブサイト iGenome からダウンロードできます。詳しくはこちら。http://cufflinks.cbcb.umd.edu/igenomes.html
この計算はサンプル数に応じてメモリを大量に消費します。Linux や Mac の場合は、top コマンドで確認してください。
1 2 3 | |
results ディレクトリに様々なファイルが出力されているのがわかると思います。
発現量解析
cummeRbund を利用して様々な発現量データ解析を行います。
まず、fastcluster と ggplot2 パッケージをインストールします。
install.packages("fastcluster")##
## The downloaded binary packages are in
## /var/folders/cz/cny0ysmx205dnj0y2_34k8cc0000gn/T//RtmpqtWkQX/downloaded_packagesinstall.packages("ggplot2")##
## The downloaded binary packages are in
## /var/folders/cz/cny0ysmx205dnj0y2_34k8cc0000gn/T//RtmpqtWkQX/downloaded_packages次に、cummeRbund をインストールします。
1 2 | |
RStudio を利用している場合は、以下のようにします。
1
| |
RStudio の Tools -> Install Packages -> Install From: Package Archive File (*.tgz) を選択。Package Archive: で cummeRbund_1.2.0.tgz を指定して、Install をクリックします。
発現データの読み込み
まず cummeRbund を使って、R に cuffdiff で定量した発現量を読み込みます。cuffdiff の結果は、outdir に保存されているはずです。そこにあるファイルを読み込んで、cuff.db というファイルに保存します。
1 2 3 4 5 | |
今回は、すでに cuff.db を作ってありますので、それを利用します。cuff.db の場所は、system.file で調べることができます。
library("cummeRbund")## Loading required package: RSQLite## Loading required package: DBI## Loading required package: ggplot2## Loading required package: reshape2## Loading required package: fastcluster## Attaching package: 'fastcluster'## The following object(s) are masked from 'package:stats':
##
## hclust## Attaching package: 'cummeRbund'## The following object(s) are masked from 'package:plyr':
##
## countdir <- system.file("extdata", package = "cummeRbund")
cuff <- readCufflinks(dir)
is(cuff)## [1] "CuffSet"cuff## CuffSet instance with:
## 3 samples
## 400 genes
## 1203 isoforms
## 662 TSS
## 906 CDS
## 1062 promoters
## 1986 splicing
## 990 relCDSここでは、dir に入っている cuffdiff の結果を読み込み、cuff.db というファイルを生成します。すでにある場合は、それを読み込んで、Rのワークスペース上に、CuffSet オブジェクトを生成します。このオブジェクトに、いろいろな操作をすることで、データを取り出したり、統計計算を行います。
まず、この CuffSet オブジェクトから、遺伝子ごとの発現量の情報を取り出します。CuffSet にはアイソフォームやプロモーターの情報も含まれています。ここでは、遺伝子ごとのデータを選択的に取り出します。
my.genes <- genes(cuff)
is(my.genes)## [1] "CuffData"my.genes## CuffData instance with:
## 400 features and 3 samplesこれによって、my.genes として CuffData オブジェクトが生成されました。3 samples で遺伝子が、400 個となっています。これはデモデータなので少ないですが、通常、マウスの場合は、3万程度になるはずです。
グローバルな統計情報
実験が上手くいっているかを確認するため、発現量のグローバルな統計情報をプロットします。いわゆる実験の quality control/check を行います。
まずは dinsity plot で発現量の分布を確認します。
dens <- csDensity(my.genes)
print(dens)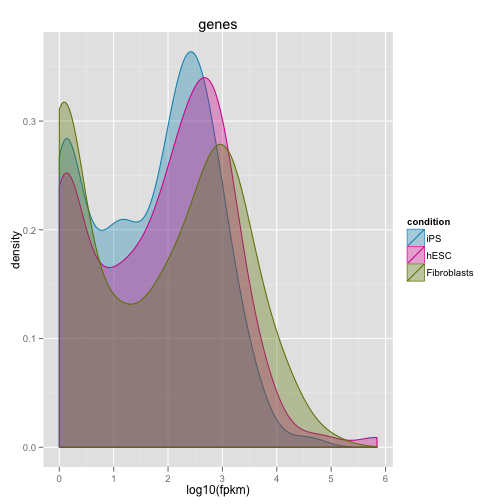
サンプルを平均化せずにひとつずつ表示するのが重要です。
dens.rep <- csDensity(my.genes, replicates = T)
print(dens.rep)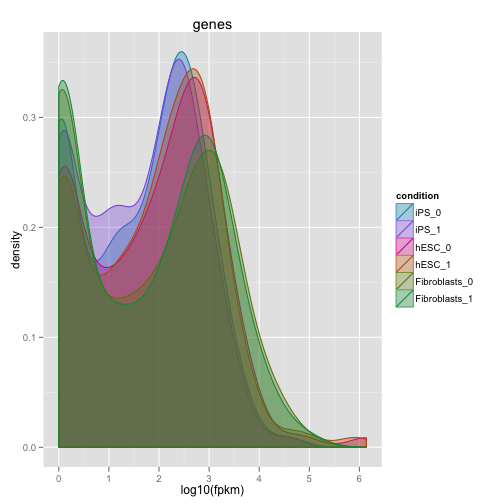
density plot では平均や分散の情報を人間が印象で評価することになります。より定量的に判断できるよう boxplot を描きます。
boxp <- csBoxplot(my.genes)
print(boxp)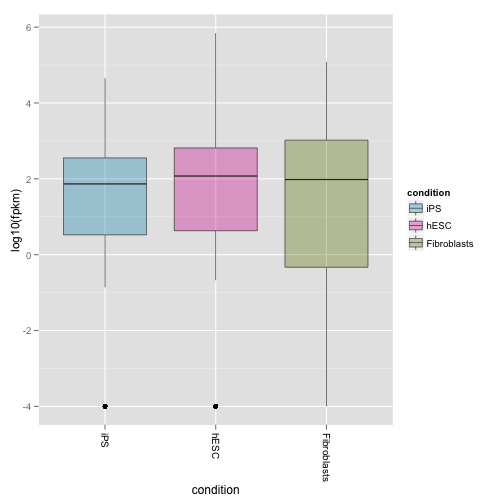
サンプル平均はなく、すべてのサンプルひとつずつ表示すするには、replicates パラメータを TRUE にします。
boxp.rep <- csBoxplot(my.genes, replicates = T)
print(boxp.rep)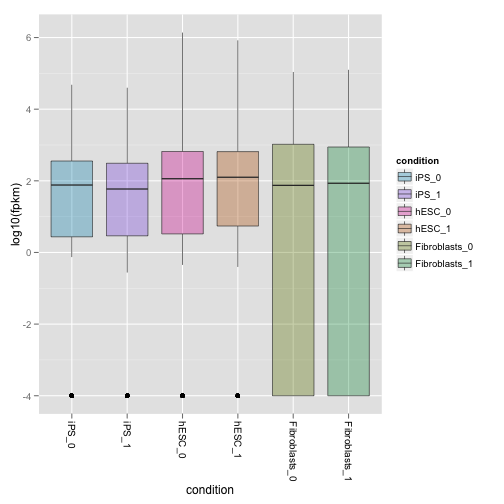
ここまでは技術的に実験が上手く行われているかを評価してきましたが、この情報が生物学的な差を捉えているかを調べます。もっとも簡単な方法は、サンプル間クラスタリングを行うことです。
dend <- csDendro(my.genes)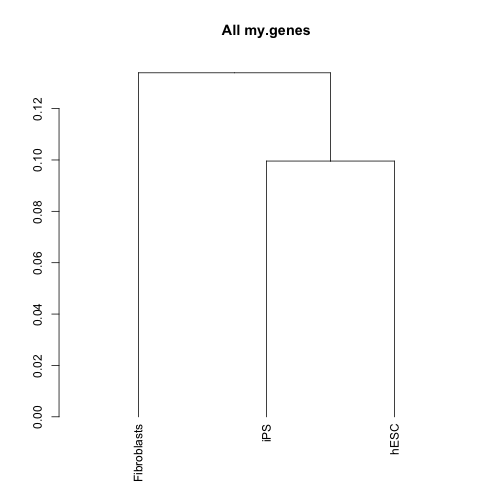
print(dend)## 'dendrogram' with 2 branches and 3 members total, at height 0.1339これによって、生物学的に同じ由来のサンプルが、同じクラスタに含まれるか、あるいは、似たサンプルが近いクラスタになるかを判断します。ここで技術的な差を越えて、生物学的に意味のあるクラスタリングをされない場合は、その後の解析に意味がないので、実験を見直す必要があります。
最後に、異なるサンプルのスキャッタープロットを描きます。
scatter <- csScatter(my.genes, "hESC", "Fibroblasts", smooth = T)
print(scatter)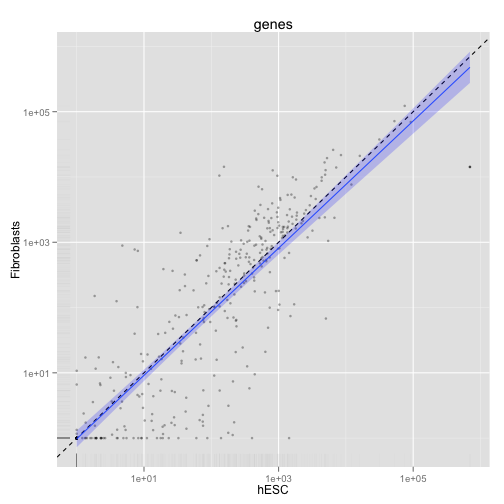
ローカルなポジティブ・ネガティブコントロールの確認
グローバルに実験結果の評価を行いましたが、技術的、あるいは、生物学的なネガティブコントロール、ポジティブコントロールの発現量を確認し、技術的、生物学的に実験の評価を行うことが重要です。RNA-seq を行う前に、そのサンプルの質を評価するため、コントロールとなる遺伝子の qPCR を行っているはずです。これらの遺伝子について、RNA-seq による発現量を確認し、実験の質を確認します。もし、qPCRのデータがない場合は、測定するか、それが可能ではない場合は、残念ですが、その RNA-seq データは計画不足として破棄するべきでしょう(ライブラリDNAの質をチェックし、シーケンスデータとの関係を示すだけであれば、ライブラリDNAに対して qPCR をする方法もありますが、ライブラリ作成前のサンプルの質のチェックとは異なります。ライブラリ作成時にPCRのステップがあることに注意してください)。
まず、ポジネガコントロール遺伝子のリストを作り、その遺伝子に対応する発現量を得ます。
data(sampleData)
my.geneset.ids <- sampleIDs
is(my.geneset.ids)## [1] "character" "vector" "data.frameRowLabels"
## [4] "SuperClassMethod"my.geneset.ids## [1] "XLOC_001363" "XLOC_001297" "XLOC_001339" "XLOC_000132" "XLOC_001265"
## [6] "XLOC_000151" "XLOC_001359" "XLOC_000069" "XLOC_000170" "XLOC_000105"
## [11] "XLOC_001262" "XLOC_001348" "XLOC_001411" "XLOC_001369" "XLOC_000158"
## [16] "XLOC_001370" "XLOC_001263" "XLOC_000115" "XLOC_000089" "XLOC_001240"my.geneset <- getGenes(cuff, my.geneset.ids)
is(my.geneset)## [1] "CuffGeneSet" "CuffFeatureSet"my.geneset## CuffGeneSet instance for 20 genes
##
## Slots:
## annotation
## fpkm
## repFpkm
## diff
## count
## isoforms CuffFeatureSet instance of size 45
## TSS CuffFeatureSet instance of size 23
## CDS CuffFeatureSet instance of size 36
## promoters CuffFeatureSet instance of size 20
## splicing CuffFeatureSet instance of size 23
## relCDS CuffFeatureSet instance of size 20取り出された my.geneset は CuffGeneSet オブジェクトとして取り出されます。
ここで data が登場してきましたが、この関数はパッケージが持つデモデータを読み込む関数です。ここでは、サンプルデータとそのID, この場合は、sampleIDs という変数を読み込んでいます。
まず、選択した遺伝子のみでサンプル間クラスタリングをすることで、サンプルを正しく分けることができるかを確認します。ここで正しく分類できない場合は、その後の解析をする意味はあまりありませんので、実験の見直しを行います。
den <- csDendro(my.geneset)## Using tracking_id, sample_name as id variables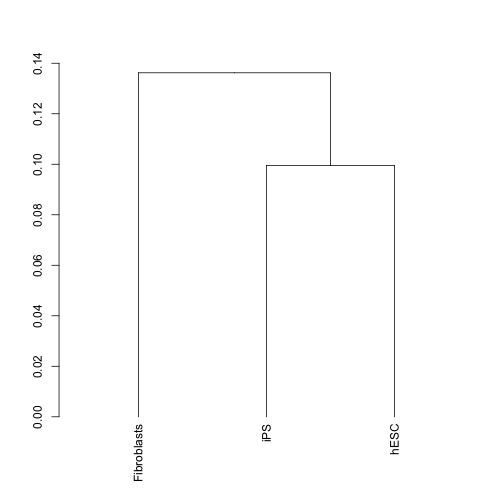
次にそれらの遺伝子についてヒートマップを描き発現量のサンプル間での違いを定性的に確認します。
h <- csHeatmap(my.geneset, cluster = "both")## Using tracking_id, sample_name as id variables## Using as id variablesprint(h)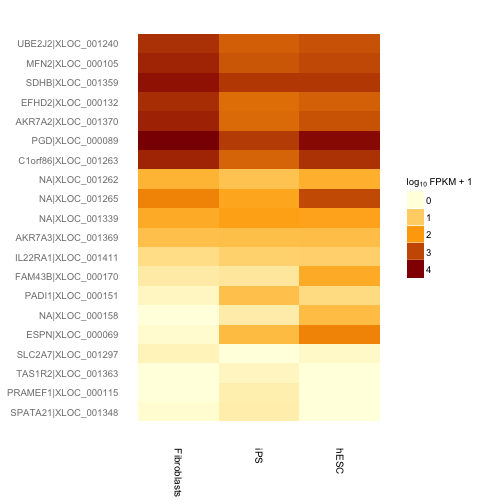
h.rep <- csHeatmap(my.geneset, cluster = "both", replicates = T)## Using tracking_id, rep_name as id variables## Using as id variablesprint(h.rep)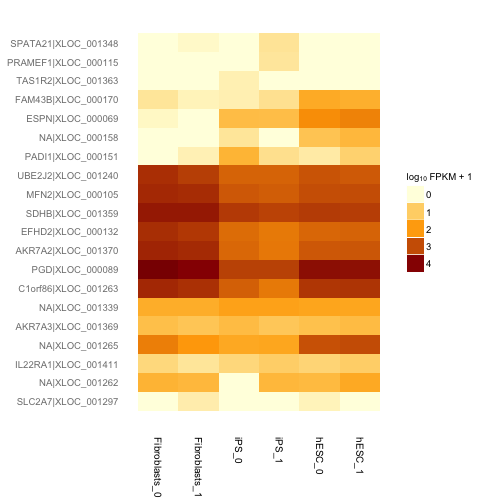
これらの遺伝子の発現量がどの程度異なるかより定量的に評価するため、barplot を描きます。
bar <- expressionBarplot(my.geneset)
print(bar)## ymax not defined: adjusting position using y instead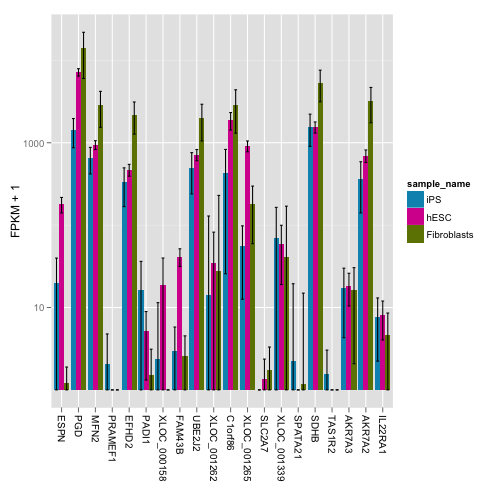
また発現量がどの程度異なるかを定量的に把握するためには、dot plot も有効です。
gl <- expressionPlot(my.geneset)
print(gl)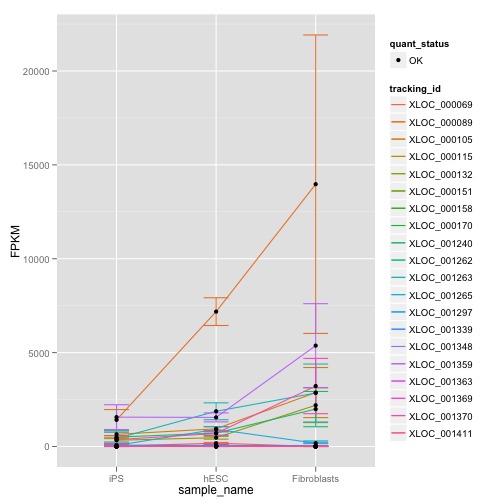
gl.rep <- expressionPlot(my.geneset, replicates = TRUE)
print(gl.rep)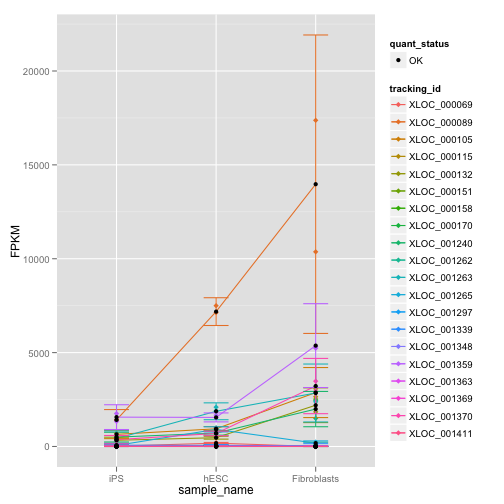
より高度なデータ探索
以上で紹介した実験とデータの質の確認を行った後、このデータから必要なデータを検索し、知識を引き出します。
まず、サンプル間で有意な遺伝子発現の差を持つ遺伝子のリストを得てみましょう。
my.sig.genes <- getSig(cuff, alpha = 0.05, level = "genes")
my.sig.table <- getSigTable(cuff, alpha = 0.01, level = "genes")## Using testResult as value column: use value.var to override.is(my.sig.genes)## [1] "character" "vector" "data.frameRowLabels"
## [4] "SuperClassMethod"head(my.sig.genes)## [1] "XLOC_000004" "XLOC_000005" "XLOC_000008" "XLOC_000009" "XLOC_000011"
## [6] "XLOC_000013"is(my.sig.table)## [1] "matrix" "array" "structure" "vector"head(my.sig.table)## hESCvsFibroblasts iPSvsFibroblasts iPSvshESC
## XLOC_000005 1 1 1
## XLOC_000008 0 1 1
## XLOC_000009 1 1 1
## XLOC_000011 0 1 1
## XLOC_000013 0 1 0
## XLOC_000014 1 NA 0発現が異なる遺伝子をどのように取り出しているかは、(How does Cuffdiff test for differential expression and regulation?)[http://cufflinks.cbcb.umd.edu/howitworks.html#diff] を参照してください。出力される p-value は、ボンフェローニ法によって多重検定の補正がされています。
my.sig.genes が data frame, my.sig.table が matrix であることに注意しましょう。
次に、発現パターンが似ているもの分類しています。ここでは k-means を利用して、4つのクラスタに分類します。
k.means <- csCluster(my.geneset, k = 4)## Using tracking_id, sample_name as id variablesk.means.plot <- csClusterPlot(k.means)
print(k.means.plot)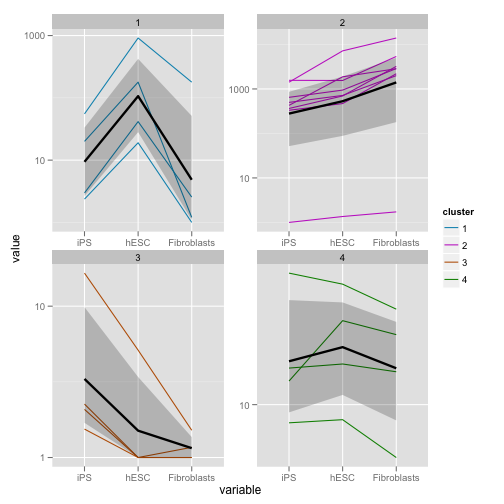
k-means はクラスタの数をユーザに与える必要があります。クラスタ数を推定したい場合は、別の適切な方法を選択する必要があります。Kを予測するのが目的ならば、混合分布のパラメータ推定を行う必要があるでしょう。またそれぞれのクラスタの分散が同じという仮定があります。それが仮定できない場合は、k-means を使うべきではありません。
次にサンプル特異的に発現している遺伝子を発見します。
my.geneset.spec <- csSpecificity(my.geneset)## Using tracking_id, sample_name as id variableshead(my.geneset.spec)## iPS_spec hESC_spec Fibroblasts_spec
## XLOC_000069 0.6382 0.7411 0.4730
## XLOC_000089 0.6074 0.6332 0.6436
## XLOC_000105 0.6163 0.6235 0.6446
## XLOC_000115 1.0000 0.4513 0.4513
## XLOC_000132 0.6127 0.6200 0.6515
## XLOC_000151 0.7245 0.6295 0.5141ここでは、各サンプルの発現量分布間の Jensen-Shannon divergence (JSD) を計算しています。JSD は分布間がどのぐらい違っているかを測る(疑)距離である、Kullback-Leibler divergence (KLD) を対称化した統計量です。ここでは、log10+1 した発現量に対して、1 - JSD を計算して出力しているので、数値が1に近付くと特異的発現をしていると言えます。
ある遺伝子と似た発現を示す遺伝子(共発現)を検索するには以下のようにします。
my.similar <- findSimilar(cuff, "PINK1", n = 20)## Using tracking_id, sample_name as id variablesis(my.similar)## [1] "CuffGeneSet" "CuffFeatureSet"my.similar## CuffGeneSet instance for 20 genes
##
## Slots:
## annotation
## fpkm
## repFpkm
## diff
## count
## isoforms CuffFeatureSet instance of size 52
## TSS CuffFeatureSet instance of size 29
## CDS CuffFeatureSet instance of size 45
## promoters CuffFeatureSet instance of size 20
## splicing CuffFeatureSet instance of size 29
## relCDS CuffFeatureSet instance of size 20ここでは、クエリとなった遺伝子の発現量とほかの遺伝子との発現分布間の JSD を計算し、JSD が小さいものをリストアップし、dot plot を描きます。
my.similar.expression <- expressionPlot(my.similar, logMode = T, showErrorbars = F)
is(my.similar.expression)## [1] "ggplot"my.similar.expression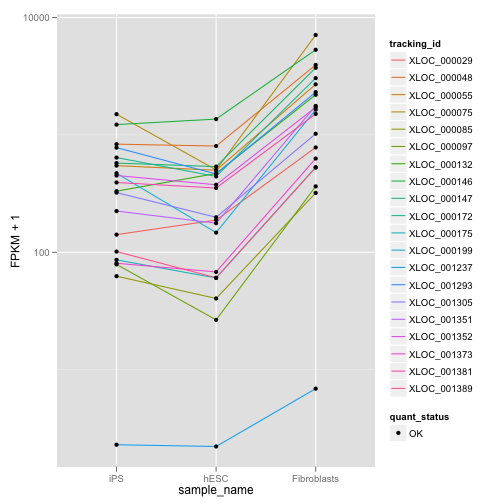
my.similar が CuffGeneSet, CuffFeatureSet オブジェクトであることに注意しましょう。
print(my.similar.expression)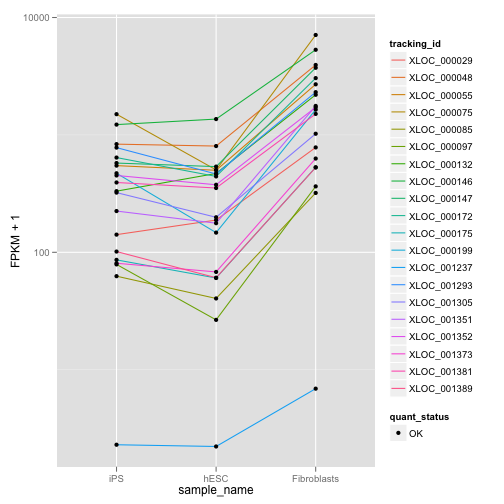
my.profile <- c(500, 0, 400)
my.similar.expression.2 <- expressionPlot(my.similar, logMode = T, showErrorbars = F)
print(my.similar.expression.2)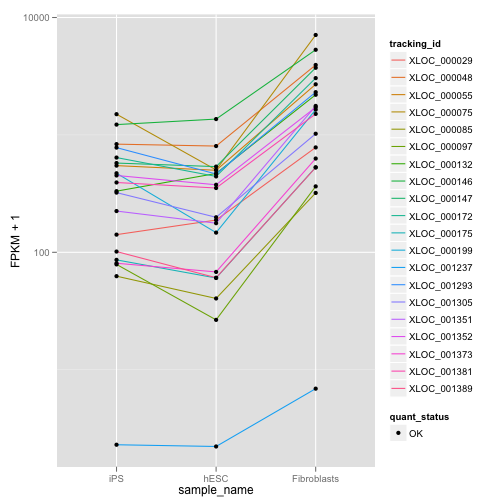
すべての遺伝子の発現量のテーブルを作成したい場合は以下のようにします。
fpkm <- fpkmMatrix(my.genes)
is(fpkm)## [1] "data.frame" "list" "oldClass" "vector"head(fpkm)## iPS hESC Fibroblasts
## XLOC_000001 20.218 3.474e-01 16.15
## XLOC_000002 0.000 0.000e+00 0.00
## XLOC_000003 0.000 0.000e+00 0.00
## XLOC_000004 0.000 6.973e+05 14237.70
## XLOC_000005 355.823 6.967e+02 48.06
## XLOC_000006 1.514 0.000e+00 0.00fpkm.log10 <- log10(fpkm + 1)
head(fpkm.log10)## iPS hESC Fibroblasts
## XLOC_000001 1.3267 0.1295 1.234
## XLOC_000002 0.0000 0.0000 0.000
## XLOC_000003 0.0000 0.0000 0.000
## XLOC_000004 0.0000 5.8434 4.153
## XLOC_000005 2.5525 2.8437 1.691
## XLOC_000006 0.4004 0.0000 0.000これを、write.table などでファイルに書き出せば、Excel やほかのソフトで読み込むことができます。
次になにを学べばよいか
シーケンサから出力される fastq にはシーケンスアダプタのコンタミネーションや、質の低いリードが含まれます。これらをフィルタリングすることは、シーケンスライブラリの質を判断したり、計算量を節約したり、その後の解析の精度を上げます。ここでは、シーケンスのフィルタリングについては紹介していませんが、重要なステップです。
rRNA のコンタミ率を計算するのも重要です。通常は、細胞のなかに含まれる total RNAのうち、mRNA は 0.01% 程度しか含まれていません。残りはほぼ rRNA です。よって、rRNA をビーズ精製など実験的手法を用いて除いていますが、このステップの精度によって、rRNA のコンタミ率が変わります。rRNA の数が多いと、RNA 由来のリードが増えてしまい、mRNA 由来の有効な read 数が相対的に減ってしまいます。リード数が減ると発見できる遺伝子の数や、捉えられる発現量のダイナミックレンジが低くなります。そこで配列解析により rRNA の量がどのぐらいあったのかを定量し、実験へフィードバックするのは大切なことです。
またマッピング後にも、マッピングスコアを用いて、マッピングの質が良くないリードを除くこともよく行われます。さらに複数箇所にマッピングされるリードを除いてしまうのか、採用するのかについても、諸説あります。基本的には、もっともスコアの高いマッピングスコアを持つリードを採用するか、ある一定の数まで重複を許す方法のいずかが採用され、後者のほうが発現定量が正確になりやすいという報告があります。
マウスやヒトなどの哺乳類ではゲノムの半分がDNAリピート関連領域だと言われています。このような領域にマップされるリードは、どのゲノム座標から得られたリードかを予測するのは難しいため、最初から、リピート領域にマップされたリードを解析の対象としない場合もあります。
発現量の定量法には、ここで利用した、FPKM だけではなく、RPM, RPKM, raw tag counts などがあります。実験デザインや発現差解析の手法によって、どの発現定量法を使うのがよいのか、いまだに議論が絶えません。最終的には、それぞれの手法を理解し試した後、qPCR や in situ hybridiation, northen blot など他の実験手法と比較してみて、自分の実験系に相応わしいものを選択するのが大切でしょう。
発現に差がある遺伝子を取り出した後、それらの遺伝子がどのようなものを含むのかを検討することも大切です。そのためには遺伝子機能の用語集である、Gene ontology が頻出するかどうかを検討したり、特定のシグナル伝達系や代謝パスウェイに関わる遺伝子が頻出するかを解析することも頻繁に行われます。
ここに挙げたような RNA-seq の諸問題にアプローチするには、今回利用した、fastq, bam file などのファイル形式を自由に操作できる技術が必要になるでしょう。また、解析の流れのどこで、どのぐらいのリードがあるのか、などの統計情報を定量したりする必要もありますが、これにもファイル操作が必要になります。R と Bioconductor を用いることで、これらのファイル操作も、比較的簡便にアプローチすることができます。それらを学び、パイプラインに組み込むことで、より正確なデータ解析ができるパイプラインができるでしょう。
参考URL
実行環境
sessionInfo()## R version 2.15.1 (2012-06-22)
## Platform: x86_64-apple-darwin9.8.0/x86_64 (64-bit)
##
## locale:
## [1] ja_JP.UTF-8/ja_JP.UTF-8/ja_JP.UTF-8/C/ja_JP.UTF-8/ja_JP.UTF-8
##
## attached base packages:
## [1] splines datasets utils stats graphics grDevices methods
## [8] base
##
## other attached packages:
## [1] Hmisc_3.9-3 survival_2.36-14 cluster_1.14.2
## [4] cummeRbund_1.99.2 fastcluster_1.1.6 reshape2_1.2.1
## [7] ggplot2_0.9.1 RSQLite_0.11.1 DBI_0.2-5
## [10] knitr_0.7.6 stringr_0.6.1 RColorBrewer_1.0-5
## [13] MASS_7.3-20 plyr_1.7.1 BiocInstaller_1.4.7
##
## loaded via a namespace (and not attached):
## [1] colorspace_1.1-1 dichromat_1.2-4 digest_0.5.2 evaluate_0.4.2
## [5] formatR_0.6 grid_2.15.1 labeling_0.1 lattice_0.20-10
## [9] memoise_0.1 munsell_0.3 proto_0.3-9.2 scales_0.2.1
## [13] tools_2.15.1