
Learning R
RとBioconductorでNGS解析: 1限 Rの基礎
はじめに
この文章は統合データベース講習会:AJACSみちのく2「RとBioconductorを使ったNGS解析」1限目「Rの基礎」講義資料です。
この文章の著作権は二階堂愛にあります。ファイルのダウンロード、印刷、複製、大量の印刷は自由におこなってよいです。企業、アカデミアに関わらず講義や勉強会で配布してもよいです。ただし販売したり営利目的の集まりで使用してはいけません。ここで許可した行為について二階堂愛に連絡や報告する必要はありません。常に最新版を配布したいのでネット上での再配布や転載は禁止します。ネット上でのリンクはご自由にどうぞ。内容についての問い合わせはお気軽にメールしてください。
対象
ここでは、本講義を理解する上で知っておきたい、Rの基本的な使いかたについて解説します。対象とするのはプログラミング初心者でRを利用したことがない人が対象です。また、なんとなくRを覚えてきた人が基礎的な知識を整理するためにも役に立つでしょう。
準備
本講義はハンズオンではなく講師の操作を眺めるスタイルですが、一緒に実行してみたい場合は、ご自分のパソコンを持ち込み、以下の準備をしてくることを強くお勧めします。推奨環境は Mac OS X や Linux ですが、Rで実行するプログラムはすべて Windows でも同じように動作します。
まずは R をインストールします。http://rstudio.org/ から、利用しているOSに適した RStudio Desktop をインストールします。起動や終了は普通のアプリケーションと同じです。起動したときに3つのペイン(ウィンドウ内にある小窓)が見えますが、左下の console という部分に、Rのプログラムを書き込んでいきます。
Proxyの設定
もし http_proxy の設定が必要な場合は、以下のように設定します。
まず Proxy の設定が必要かどうかは以下のコマンドが正常に終了するかどうかで判断できます。
source("http://bioconductor.org/biocLite.R")正常に終了しない場合は、~/.Renviron というファイルを作成し中身を
1
| |
とします。proxyserverr, prot には、それぞれの機関で提供されている Proxy サーバのアドレスとポート番号を入力します。その後、RStudio を再起動してください。
さらにRのコンソールに
1
| |
を入力します。
R の動作確認
試しに、以下のように入力して同じ結果になれば準備は終了です。ただし、あなたの環境では ## は表示されませんが、それで正常です。この資料では、## が付いていない行が、あなたが入力するコマンドです。## が付いている行は R が出力する結果なので、あなたが入力することはありません。
1 + 1 # 足し算## [1] 22 * 2 # かけ算## [1] 415 / 3 # 割り算## [1] 517 %% 3 # 余り## [1] 22^2 # べき乗## [1] 4sqrt(2) # 平方根## [1] 1.414pi # $\pi$## [1] 3.142bmi <- 57.8 / 1.63^2 # Body Mass Index
bmi## [1] 21.75学習範囲
Rを含む、プログラミング言語を理解するときに必要なことは、以下の3つです。
- データ型とデータ構造を知る
- 基本の3つの処理の書きかたを理解する
- 自分が使う便利な機能(関数、パッケージ)を覚える
この文章では、この3つについて簡単に説明します。また、バイオインフォマティクス向けの機能を集めた Bioconductor というソフトウェアパッケージについても紹介します。
データ型
データ型 (data type) とはデータの形式のことを意味します。例えば数値や文字列、論理型などがあります。
x <- c()
x## NULLy <- c(1, 2)
y## [1] 1 2z <- c("ATG", "GCG", "CTG", "TTA")
z## [1] "ATG" "GCG" "CTG" "TTA"b <- c(TRUE, FALSE)
b## [1] TRUE FALSEデータ型の変換
異なるデータ型を行き来したい場合があります。多くのプログラムのエラーはここが原因となるため、データの型の変換方法と、型を調べる方法を理解することは重要です。
y <- as.character(y)
y## [1] "1" "2"as.numeric(y)## [1] 1 2y## [1] "1" "2"型の確認
mode(1)## [1] "numeric"mode("A")## [1] "character"is(1)## [1] "numeric" "vector"is("1")## [1] "character" "vector" "data.frameRowLabels"
## [4] "SuperClassMethod"データ構造
前項ではデータを格納する型について解説しました。このデータを集めて、決まった構造でデータを束ねておくのがデータ構造 (data structure) です。プログラミング言語が、決まったデータ構造を提供することで、様々な計算が楽にできるようになります。なぜなら、データの中身が違っても構造が同じことを利用し、汎用的な計算機能(関数)を用意できるからです。また、言語側から考えると、データ構造に対する計算を最適化しておくことができ、ユーザはそのデータ構造さえ使っておけば、高速化などに配慮しなくても良くなります。プログラム言語の善し悪しはこのデータ構造とそれを処理する機能の善し悪しによって決まるとも言えます。Rでは統計解析に向いた様々なデータ構造が用意されています。ここではそのなかでもよく使うデータ構造、vector, list, matrix, data frame について述べます。
vector
vector は「1種類のデータ型」からなる「1次元データ」を格納するためのデータ構造です。
x <- c(3, 2, 4, 1)
x## [1] 3 2 4 1ここでは、3, 2, 4, 1という1次元の数列を生成して、xという変数に格納します。変数とは、生成したデータを保存しておくためのものです。データ型、データ構造、変数の関係が混乱しないよう整理してみてください。この例では、変数が x で、データ構造が vector, データ型は numeric です。
list
list は「複数のデータ型」からなる「1次元データ」を格納することができるデータ構造である。例えば文字列型と数値を同時に格納できます。
y <- list(3, 2, 4, 1)
y## [[1]]
## [1] 3
##
## [[2]]
## [1] 2
##
## [[3]]
## [1] 4
##
## [[4]]
## [1] 1この例では、変数が y で、データ構造が list, データ型は numeric です。
(発展: リストは1次元データを格納しますが、様々なデータを持てるので、要素に vector も持つことができます。)
matrix
matrix は「1種類のデータ型」からなる「2次元データ」を格納するためのデータ構造である。いわゆる行列を表現することができます。
z <- matrix(c(3, 2, 1, 2, 0, 1, 4, 5, 7, 1, 3, 3), nrow = 3)
z## [,1] [,2] [,3] [,4]
## [1,] 3 2 4 1
## [2,] 2 0 5 3
## [3,] 1 1 7 3この例では、変数が z で、データ構造が matrix, データ型は numeric です。
data frame
data frame は「複数のデータ型」からなる「2次元データ」を格納するためのデータ構造です。Excel などのスプレットシートをイメージすればよいでしょう。数値行列だけを持つ場合は、matrix のほうが data frame より高速です。
(発展: data.frame の文字列の列は、factor というデータ型の場合があります。factor 型を使えば、統計で言うところのカテゴリカル変数を表現できます。)
d <- data.frame(first = c(3, 2, 1), first.base = c("A", "T", "G"), second = c(4,
5, 7), second.base = c("C", "G", "C"))
d## first first.base second second.base
## 1 3 A 4 C
## 2 2 T 5 G
## 3 1 G 7 Cこの例では、変数が d で、データ構造が data frame, データ型は numeric と character です。
データ構造を確認する
どんなデータ構造を利用しているかを確認する方法を述べます。自分が使っているデータ構造を理解しておかないと、プログラムのエラー(バグ)の原因となります。特定のデータ構造を前提としている機能を使うとき、データ構造や型が異なる場合は、自動的に型や構造を変換したり、エラーを出したりします。
is(z)## [1] "matrix" "array" "structure" "vector"is(d)## [1] "data.frame" "list" "oldClass" "vector"str(d)## 'data.frame': 3 obs. of 4 variables:
## $ first : num 3 2 1
## $ first.base : Factor w/ 3 levels "A","G","T": 1 3 2
## $ second : num 4 5 7
## $ second.base: Factor w/ 2 levels "C","G": 1 2 1データにアクセスする
データ構造にデータを出し入れする方法を示します。行や列の各要素を示す番号を添字 (index) と呼びます。これを使ってデータを取り出したり、代入することが可能です。
まず vector の要素にアクセスする方法を示します。添字は一番左から1,2,3,… となります。プログラミング言語によっては、添字が 0 から始まるものもありますが、R は 1 であることに注意しましょう。
x <- c(3, 2, 4, 1)
x[1]## [1] 3x[1] <- 4
x## [1] 4 2 4 1x[1:2]## [1] 4 2x[1:2] <- c(3, 8)
x## [1] 3 8 4 1次に list の要素にアクセスする方法を示します。
x <- list(3, 2, 4, 1)
x[[1]]## [1] 3x[[2]] <- 5
x## [[1]]
## [1] 3
##
## [[2]]
## [1] 5
##
## [[3]]
## [1] 4
##
## [[4]]
## [1] 1次は、matrix の要素にアクセスする方法を示します。横方向が行、縦方向が列であり、これは数学の行列と同じです。行と列の方向は漢字の「作り」の最初の画の方向と覚えます。
x <- matrix(c(3, 2, 1, 2, 0, 1, 4, 5, 7, 1, 3, 3), nrow = 3)
x[1, ] # 1行目にアクセス## [1] 3 2 4 1x[, 2] # 2列目にアクセス## [1] 2 0 1x[3, 2] # 3行2列目にアクセス## [1] 1x[3, 2] <- 9 # 3行2列目に9を
x[3, ]## [1] 1 9 7 3最後に data frame の要素にアクセスする方法を示します。
x <- data.frame(first = c(3, 2, 1), first.base = c("A", "T", "G"), second = c(4,
5, 7), second.base = c("C", "G", "C"))
x$first## [1] 3 2 1x$first.base## [1] A T G
## Levels: A G Tx$first.base <- c("T", "C", "A")
x$first.base## [1] "T" "C" "A"ワークスペースと作業ディレクトリ
前項で作った変数は、ワークスペースと呼ばれるメモリ上の作業空間に保存されています。よってRを終了するまでメモリ上に保存されていますが、ハードディスクに書き込まれているわけではないので、Rを終了すると消えてしまいます。
解説: メモリとは計算機の頭脳であるCPUがアクセスする記憶装置です。電源を切ったりプログラムが終了するとメモリ内の情報は消えてしまいます。記憶が残るようにするには、ハードディスク(HDD)や、フラッシュメモリ(ソリッドステートドライブ, SSDなど)という装置に保存する必要があります。メモリはHDDやSDDに比べて、データの出し入れが高速なため、計算に必要なデータはいったん、HDDやSSDなどの記憶装置からメモリに読み出して、計算するのが普通です。Rも変数に格納されたデータやコマンド履歴や作成した関数などはメモリ上に蓄えられています。これらをR終了後にも再利用したい場合は、HDDやSSDに書き込む必要があります。
現在のワークスペースの中身を見るには、
ls()## [1] "ans" "b"
## [3] "bar" "bmi"
## [5] "boxp" "boxp.rep"
## [7] "complement" "cuff"
## [9] "d" "den"
## [11] "dend" "dens"
## [13] "dens.rep" "dir"
## [15] "even" "fpkm"
## [17] "fpkm.log10" "gene"
## [19] "gene.lm" "genome.length.file"
## [21] "gl" "gl.rep"
## [23] "h" "h.rep"
## [25] "i" "ids"
## [27] "jaspar" "jaspar.scores"
## [29] "k.means" "k.means.plot"
## [31] "KnitPost" "mat.cor"
## [33] "mat.cor.max" "mat.cor.min"
## [35] "my.esets" "my.genes"
## [37] "my.geneset" "my.geneset.ids"
## [39] "my.geneset.spec" "my.profile"
## [41] "my.sig.genes" "my.sig.table"
## [43] "my.similar" "my.similar.expression"
## [45] "my.similar.expression.2" "nanog.bed.file"
## [47] "nanog.bin500" "nanog.cov"
## [49] "nanog.df" "nanog.fat"
## [51] "nanog.gr" "nanog.unistd"
## [53] "num" "oct4.anno"
## [55] "oct4.anno.table" "oct4.bed.file"
## [57] "oct4.bin500" "oct4.bp.goterm"
## [59] "oct4.cc.goterm" "oct4.cov"
## [61] "oct4.df" "oct4.fat"
## [63] "oct4.go" "oct4.gr"
## [65] "oct4.jaspar" "oct4.mf.goterm"
## [67] "oct4.motif" "oct4.motif.pwms"
## [69] "oct4.peaksWithSeqs" "oct4.seqs"
## [71] "oct4.unistd" "odd"
## [73] "path" "PINK1"
## [75] "quga.oct4.nanog" "quga.oct4.sox2"
## [77] "quga.sox2.nanog" "sampleGeneSet"
## [79] "sampleIDs" "scatter"
## [81] "sox2.bed.file" "sox2.bin500"
## [83] "sox2.cov" "sox2.df"
## [85] "sox2.fat" "sox2.gr"
## [87] "sox2.unistd" "TSS.mouse.NCBIM37"
## [89] "x" "x.mean"
## [91] "x.sd" "x.sum"
## [93] "xm" "y"
## [95] "z"とします。
このワークスペースをハードディスクに書き込んで、次回 R が起動したときに、まったく同じワークスペースを再現することができます。
save.image()また、Rを終了するコマンドを入力するとワークスペースを保存するか聞かれますので、これに y と答えても同じ意味になります。
1 2 | |
このファイルはどこに保存されているのでしょうか? これを知るにはまず作業ディレクトリについて知る必要があります。作業ディレクトリとは、Rで作業しているときに、ハードディスク上で基点としている場所のことです。これを知るには、
getwd()## [1] "/Users/itoshi/Desktop/20120824_RBioCNGS_TohokuU/NGS_analysis_with_R_Bioconductor/doc"とします。
作業ディレクトリを変更したい場合は、
1
| |
とします。
q や save.image で保存されるファイルは、この作業ディレクトリ上に、.RData というファイル名で保存されます。(ファイル名がピリオドで始まるファイルを、dot file あるいは、隠しファイルと呼び、Windows や Mac では標準の設定では、見ることはできません)
ファイル名を指定して、ワークスペースを保存したい場合は、
1
| |
とします。R では ファイル名.rda という拡張子がよく用いられます。
3つの基本的な処理
プログラミング言語は基本的には3つの処理の組み合わせで実行されます。ダイクストラの構造化プログラミング (Structured Programming) では、基本的な処理として3つの処理が挙げられています。
順次: 書かれた順番に処理していく 反復: 条件が満されるまで処理を繰り返す 分岐: 条件が成立する場合は実行する
順次処理については、これまでも示してきた通りなので割愛しますが、Rの場合は文単位 (行ではない) で上から下へと順次処理されます。
次に反復処理について示します。ここでは、for を利用した繰り返しの構文を紹介します。
ans <- 0
for (i in 1:10) {
ans <- ans + i
}
ans## [1] 55次に if を使って条件によって処理を分岐してみましょう。
odd <- 0
even <- 0
for (i in 1:10) {
if (i%%2 == 0) {
even <- even + i
} else {
odd <- odd + i
}
}
cat(even, odd, "\n")## 30 25関数
関数 (function) とは、便利な機能のひとかたまりにしたものです。プログラミング言語には様々な関数が定義されており、この関数の豊富さが言語の使い易さに深く関わっています。Rでは文字や数値の処理だけでなく、統計処理のための便利な関数が多数用意されています。関数は膨大にあるので、まず自分の使う関数だけを覚えておき、必要に応じてリファレンスや Web検索などで必要な関数を調べるのがお勧めです。
簡単な統計量を計算する関数を紹介します。
x <- 1:100
x.sum <- sum(x)
x.mean <- mean(x)
x.sd <- sd(x)
cat(x.sum, x.mean, x.sd, "\n")## 5050 50.5 29.01関数を自分で作ることもできます。自分がよく使う機能をまとめておくことで、プログラムの見通しもよくなったり、機能が再利用しやすくなったり、プログラムコードのメンテナンスしやすくなるなどのメリットが多いので、積極的に利用したい機能です。
関数を作るコツは、ひとつの関数はなるべくひとつの機能を果すようにし、画面1枚以内に収まる程度にすることです。れが守られないときは、よく考えて、複数の関数に分割することが重要です。
ここでは簡単な関数の例として、DNA配列の相補鎖を計算する関数を定義してみましょう。
complement <- function(x) {
if (x == "A") {
return("T")
} else if (x == "T") {
return("A")
} else if (x == "G") {
return("C")
} else if (x == "C") {
return("G")
} else {
return("N")
}
}
complement("A")## [1] "T"complement("G")## [1] "C"complement("*")## [1] "N"定義されている関数の使い型を調べるには help を利用します。Rの良いところとして、関数の多くに実行例が付属していることです。これを実行するには、example を利用します。
関数の使いかたを調べる
help(sum)
example(mean)##
## meanR> x <- c(0:10, 50)
##
## meanR> xm <- mean(x)
##
## meanR> c(xm, mean(x, trim = 0.10))
## [1] 8.75 5.50パッケージを使う
パッケージとは、複数の便利な関数をまとめて提供するものです。R のパッケージは CRAN と Bioconductor から配布されています。パッケージは、世界中の開発者によって各々、開発されており、最新の統計手法や、様々な実験機器データを扱うパッケージなど様々です。特に Bioconductor はバイオインフォマティクス関連のパッケージに特化しており、CRAN よりもソースコードのレビューやドキュメンテーションのルールが厳しく、パッケージが比較的良質に保たれています。
まず、CRANからパッケージをインストールする方法を述べます。パッケージはコマンドから簡単にインストールができ install.packages を利用します。
install.packages("ggplot2")##
## The downloaded binary packages are in
## /var/folders/cz/cny0ysmx205dnj0y2_34k8cc0000gn/T//Rtmp9ixJXF/downloaded_packagesインストールが正常に終了していれば、以下のように library でパッケージの機能を読み込むことができます。
library("ggplot2")次に、Bioconductor からパッケージをダウンロードします。これにはまず専用の関数を biocLite を設定する必要します。ここでは、biocLite を設定し、マウス脳トランスクリプトームデータ解析ツールである BrainStars パッケージをインストールしてみます。
いろいろメッセージが表示されるが気にする必要はありません。
source("http://bioconductor.org/biocLite.R")
biocLite("BrainStars")## BioC_mirror: http://bioconductor.jp/## Using R version 2.15, BiocInstaller version 1.4.7.## Warning: 開けません:HTTP ステータスは '404 Not Found' です## Warning: 開けません:HTTP ステータスは '404 Not Found' です## Warning: unable to access index for repository
## http://brainarray.mbni.med.umich.edu/bioc/bin/macosx/leopard/contrib/2.15## Installing package(s) 'BrainStars'## Warning: 開けません:HTTP ステータスは '404 Not Found' です## Warning: 開けません:HTTP ステータスは '404 Not Found' です## Warning: unable to access index for repository
## http://brainarray.mbni.med.umich.edu/bioc/bin/macosx/leopard/contrib/2.15## Warning: 開けません:HTTP ステータスは '404 Not Found' です## Warning: 開けません:HTTP ステータスは '404 Not Found' です## Warning: unable to access index for repository
## http://brainarray.mbni.med.umich.edu/bioc/bin/macosx/leopard/contrib/2.15Proxy がある場合は、以下のような設定を先にしておきます。
options(RCurlOptions = list(proxy = "proxy.l2.med.tohoku.ac.jp:8080"))インストールしたパッケージは、CRANパッケージと同様に、library で使うことができます。ここでは、3つの遺伝子IDを指定し、それらの脳50領域の遺伝子発現量を取り出しています。
library(BrainStars)
ids <- c("1439627_at", "1439631_at", "1439633_at")
my.esets <- getBrainStars(query = ids, type = "expression")
exprs(my.esets)## LS LS.1 RS RS.1 Cx_motor Cx_motor.1 Cx_cingulate
## 1439627_at 12.868 12.854 5.686 5.771 5.643 5.650 7.125
## 1439631_at 6.408 6.851 6.562 6.485 6.191 6.493 6.194
## 1439633_at 10.286 10.164 10.449 10.540 10.293 10.366 10.452
## Cx_cingulate.1 OB_anterior OB_anterior.1 OB_posterior
## 1439627_at 7.657 10.992 10.772 10.83
## 1439631_at 6.539 7.315 7.445 7.45
## 1439633_at 10.421 10.243 10.340 10.37
## OB_posterior.1 Pir Pir.1 Tu Tu.1 ventral_S ventral_S.1
## 1439627_at 10.592 6.263 5.842 8.711 8.297 5.557 5.459
## 1439631_at 7.807 7.019 6.966 6.674 6.667 6.766 6.654
## 1439633_at 10.338 10.735 10.845 9.657 9.823 10.081 9.937
## CA1 CA1.1 CA2_CA3 CA2_CA3.1 DG DG.1 A_anterior
## 1439627_at 6.477 7.560 6.044 6.419 8.213 8.600 6.147
## 1439631_at 6.469 6.562 6.759 6.314 6.796 6.843 6.815
## 1439633_at 10.611 10.637 10.267 10.221 10.948 10.956 9.842
## A_anterior.1 A_posterior A_posterior.1 GP GP.1 CPu_lateral
## 1439627_at 5.156 4.999 4.934 8.210 8.028 7.961
## 1439631_at 6.629 6.794 6.613 6.867 6.503 6.504
## 1439633_at 9.794 10.356 10.491 10.066 9.948 9.233
## CPu_lateral.1 CPu_medial CPu_medial.1 MD MD.1 VA_VL
## 1439627_at 7.956 7.665 7.469 11.754 12.097 12.284
## 1439631_at 6.536 6.417 6.654 6.467 6.543 6.735
## 1439633_at 9.171 9.408 9.245 11.452 11.459 10.871
## VA_VL.1 VPM_VPL VPM_VPL.1 LG LG.1 MG MG.1 Hb
## 1439627_at 11.720 12.228 11.945 12.383 12.309 12.130 12.040 12.943
## 1439631_at 6.323 6.121 6.524 6.234 6.738 6.432 6.439 6.882
## 1439633_at 10.761 10.867 10.775 10.611 10.463 11.028 10.946 9.832
## Hb.1 Pineal Pineal.1 M M.1 ME ME.1 Pituitary
## 1439627_at 12.993 10.500 10.418 9.154 8.963 6.477 6.633 5.038
## 1439631_at 6.917 8.658 8.619 7.096 6.844 8.122 8.344 7.431
## 1439633_at 10.133 8.645 8.950 9.524 9.837 9.260 8.916 10.298
## Pituitary.1 SCN SCN.1 MPA MPA.1 SO SO.1 Pa
## 1439627_at 4.505 11.140 10.935 10.826 11.055 10.234 10.369 10.331
## 1439631_at 8.091 7.081 7.358 6.861 6.996 6.623 6.538 6.880
## 1439633_at 10.470 8.846 8.977 9.337 9.023 9.647 9.585 9.489
## Pa.1 SPa_ventral SPa_ventral.1 SPa_dorsal SPa_dorsal.1 DM
## 1439627_at 10.196 11.342 11.038 6.941 6.890 7.063
## 1439631_at 6.978 8.023 7.141 7.994 7.283 6.834
## 1439633_at 9.548 9.548 9.513 9.484 9.703 9.304
## DM.1 VMH VMH.1 Arc Arc.1 LH LH.1 PAG PAG.1 SC
## 1439627_at 6.580 5.898 6.079 6.212 6.736 10.061 9.855 6.723 7.451 8.381
## 1439631_at 7.252 6.707 6.938 7.088 7.065 6.703 6.734 6.757 6.720 6.902
## 1439633_at 9.518 9.437 9.377 9.407 9.035 9.570 9.356 9.807 9.985 10.178
## SC.1 IC IC.1 VTA VTA.1 SN SN.1 Tg Tg.1
## 1439627_at 9.588 10.655 7.416 7.060 6.054 7.071 8.107 6.141 8.989
## 1439631_at 6.917 6.891 6.631 7.721 7.366 6.185 6.171 6.762 6.785
## 1439633_at 10.239 10.180 10.324 9.732 9.896 10.090 10.053 10.125 10.295
## Pn Pn.1 MVe MVe.1 Cb_vermis Cb_vermis.1 Cb_lobe
## 1439627_at 11.019 11.206 10.069 10.41 13.023 12.88 12.953
## 1439631_at 6.562 6.508 6.547 6.52 7.789 7.57 7.641
## 1439633_at 10.635 10.673 10.355 10.43 10.614 10.51 10.683
## Cb_lobe.1 Cb_nucleus Cb_nucleus.1 spinal_cord_anterior
## 1439627_at 13.105 9.774 10.216 7.581
## 1439631_at 7.819 6.534 6.508 6.352
## 1439633_at 10.771 10.593 10.547 9.931
## spinal_cord_anterior.1 spinal_cord_posterior
## 1439627_at 7.675 9.452
## 1439631_at 6.391 6.695
## 1439633_at 9.941 9.976
## spinal_cord_posterior.1 Retina Retina.1
## 1439627_at 9.638 8.233 6.822
## 1439631_at 6.507 7.340 6.641
## 1439633_at 10.019 8.668 8.967簡単な統計解析
Rは、統計解析のための関数を多く持つため、あらゆる統計解析が可能です。ここでは簡単な例として、検定と回帰を取り上げます。
検定
ある遺伝子の発現量を測定したとします。ここでは、処理群(ここではExp)と対照群(ここではCont)でそれぞれ n = 3, つまり3回の biological replication の発現量を測定してあるとします。これを data frame に格納し、処理、対照群間の平均値の差の検定、ここではT検定を行なってみます。
gene <- data.frame(sample = c("Exp", "Exp", "Exp", "Cont", "Cont", "Cont"),
expression = c(12.8, 13.7, 15.2, 8, 7.3, 9.9))
t.test(expression ~ sample, data = gene)##
## Welch Two Sample t-test
##
## data: expression by sample
## t = -5.26, df = 3.957, p-value = 0.006442
## alternative hypothesis: true difference in means is not equal to 0
## 95 percent confidence interval:
## -8.415 -2.585
## sample estimates:
## mean in group Cont mean in group Exp
## 8.4 13.9統計解析を行うときには 必ず グラフを書いて、データの傾向を目視することを忘れてはなりません。プログラムのバグを避けたり、データの不具合や入れ違いに気付くためには不可欠だからです。統計処理をしてしまうと、それに気付くことは非常に困難です。
ここでは boxplot で2群の傾向を可視化しています。
plot(expression ~ sample, data = gene)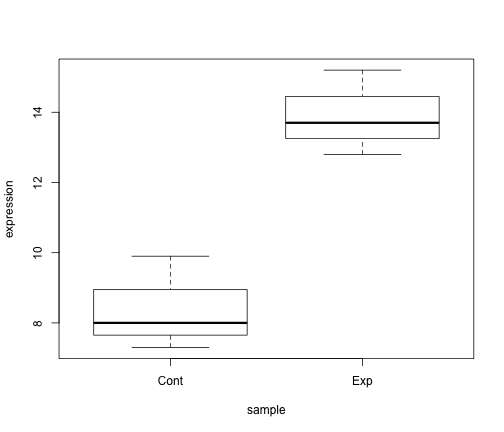
回帰
ここでは、一日ごとに遺伝子発現を測定したとしましょう。この遺伝子発現が時間に応じてどのように変化しているか、その傾向を知りたいとします。そこで、一日ごとの発現量を data frame に格納し、回帰分析を行ってみます。
gene <- data.frame(time = c(0, 1, 2, 3, 4, 5), expression = c(1.8, 3.7, 4.2,
8, 10.3, 13.9))
gene.lm <- lm(expression ~ time, data = gene)
gene.lm##
## Call:
## lm(formula = expression ~ time, data = gene)
##
## Coefficients:
## (Intercept) time
## 0.976 2.403summary(gene.lm)##
## Call:
## lm(formula = expression ~ time, data = gene)
##
## Residuals:
## 1 2 3 4 5 6
## 0.824 0.321 -1.582 -0.185 -0.288 0.910
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 0.976 0.744 1.31 0.25978
## time 2.403 0.246 9.78 0.00061
##
## Residual standard error: 1.03 on 4 degrees of freedom
## Multiple R-squared: 0.96, Adjusted R-squared: 0.95
## F-statistic: 95.6 on 1 and 4 DF, p-value: 0.000613次に生データと回帰分析によって得られた回帰直線を同時にプロットします。
plot(gene, type = "b", xlim = c(0, 6), ylim = c(0, 14))
abline(gene.lm, col = "blue")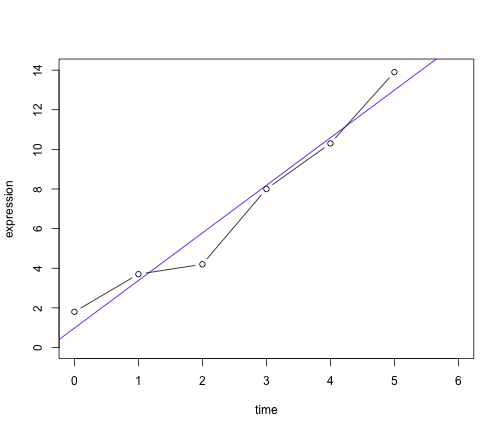
次になにを学べばよいか
何度も強調しますが、まず自分が必要な機能から学ぶのがよいです。ただし最低限Rに実験データを取り込むために、ファイルの入出力の方法を学んでおくとよいでしょう。具体的には、read.csv, read.table, write.table などです。
またデータ型やデータタイプに関しては主要なものだけを説明しており、Rの機能を網羅しているわけではありません。特に factor 型を理解しておくと良いでしょう。また、for ではなく apply 族と呼ばれる関数群を使うことや、反復処理ではなくベクトル計算を行うことなどの R らしいコーディングスタイルを学ぶことも重要です。
最終的には自分のデータに合せて、Bioconductor, CRAN パッケージの使いかたを覚えるのが次の目標となります。この講義では、RNA-seq, ChIP-seq を例に、Bioconductor の使い方を解説していきます。
練習問題
問題1
1から順に数字を表示しなさい。ただしその数が3で割り切れるならは数字のかわりに Fizz と、5で割り切れるなら Buzz と、3でも5でも割り切れる場合は FizzBuzz と表示しなさい (いわゆる FizzBuzz 問題と呼ばれており、プログラマならば、なにも参考にせずに、2分で解けなければならないと言われている)。
問題2
問題1を条件分岐を使わずに解きなさい。
問題3
以下のようにすると、Rに組み込まれているデータフレーム iris を読み込むことができる。
1
| |
この iris の1から4列の各列の標準偏差を for を使って求めなさい。
問題4
問題2で利用した iris の列ごとの平均を for を使って求めなさい。
問題5
問題2を for を用いずに一行で求めなさい。ヒント: sapply あるいは、colMeans を help で調べる。
問題6
問題2で利用した iris の1から4列の各列の標準偏差を for を用いずに、sapply 関数を使って求めなさい。
問題7
「パッケージを使う」のパートで出てきた exprs(my.esets) を boxplot 関数を用いてプロットせよ。
発展問題
ここで解説したRプログラミングの知識がすでにあるという人は、講義を聞かずに以下の問題を解いていてもよい。ウェブ検索を駆使して答えよ。
初級
- 前項の練習問題すべてを、ヘルプや検索を用いずに、20分以内に解け
- 講義資料中にある「DNA配列の相補鎖を計算する関数」を、関数を自作せずに、Rに組み込まれているたったひとつの関数で実現せよ。
- 練習問題1の結果をファイルを出力せよ。ただし一行ごとにひとつの数字または文字列を表示せよ。
中級
- lapply, apply, sapply の違いについて述べよ。
- Rに組み込みこまれている apply 族関数と plyr package で提供される関数について比較し説明せよ。
- multicore package を利用して、練習問題1をマルチコアを使った並列計算するコードに直せ。
上級
- 問題1をS3, S4, R5 オブジェクト指向で実装し直せ。
- Rのスコープについて解説せよ。ヒント: (環境、レキシカルスコープ)
- 総称関数とはなにか。summary か plot を例に説明せよ。
- 上記の回答をすべて、R markdown で執筆し、RPubs にアップロードせよ。
実行環境
sessionInfo()## R version 2.15.1 (2012-06-22)
## Platform: x86_64-apple-darwin9.8.0/x86_64 (64-bit)
##
## locale:
## [1] ja_JP.UTF-8/ja_JP.UTF-8/ja_JP.UTF-8/C/ja_JP.UTF-8/ja_JP.UTF-8
##
## attached base packages:
## [1] datasets utils stats graphics grDevices methods base
##
## other attached packages:
## [1] ggplot2_0.9.1 knitr_0.7.6 stringr_0.6.1
## [4] RColorBrewer_1.0-5 MASS_7.3-20 plyr_1.7.1
## [7] BiocInstaller_1.4.7
##
## loaded via a namespace (and not attached):
## [1] colorspace_1.1-1 dichromat_1.2-4 digest_0.5.2 evaluate_0.4.2
## [5] formatR_0.6 grid_2.15.1 labeling_0.1 memoise_0.1
## [9] munsell_0.3 proto_0.3-9.2 reshape2_1.2.1 scales_0.2.1
## [13] tools_2.15.1